
PMS2




PMS2(postmeiotic segregation increased 2)は、ヒトではPMS2遺伝子にコードされる酵素(エンドヌクレアーゼ)である[4]。
機能
[編集]PMS2遺伝子は7番染色体上にクラスターとして存在するPMS2遺伝子ファミリーの一員である。ヒトPMS2関連遺伝子はバンド7p12、7p13、7q11、7q22に位置している。これらのホモログのエクソン1から5はPMS2と高度の同一性を示す[5]。PMS2遺伝子の産物はDNAミスマッチ修復(MMR)に関与している。PMS2タンパク質はMLH1とヘテロ二量体を形成し、この複合体はMSH2関連複合体によるミスマッチ塩基や挿入/欠失ループの認識後に活性化される。PMS2遺伝子の変異は遺伝性非ポリポーシス大腸癌(HNPCC)やターコット症候群と関係している[6]。
ミスマッチ修復とエンドヌクレアーゼ活性
[編集]PMS2はミスマッチ修復に関与しており、MutLホモログに存在するmeta-binding motifの完全性に依存した潜在型エンドヌクレアーゼ活性を有することが知られている。PMS2はエンドヌクレアーゼとして、非連続的なDNA鎖にさらにニックを導入する[7]。
相互作用
[編集]PMS2はMLH1と相互者王してMutLαヘテロ二量体を形成することが示されている[8][9][10][11][12][13]。MLH1上の相互作用ドメイン(492–742番残基)をめぐって、MLH3、PMS1、PMS2の間には競合が存在する[9]。
PMS2の相互作用ドメインにはロイシンジッパータンパク質に特徴的なヘプタッドリピートが存在し、MLH1の506–756番残基と相互作用する[10]。
MutSヘテロ二量体(MutSαやMutSβ)は、ミスマッチ部位への結合に伴ってMutLαと結合する。MutLαは、ミスマッチ認識段階からその他の過程、すなわち新生DNA鎖からのミスマッチの除去、分解されたDNAの再合成、DNA中のニックの修復などへの連結を担っていると考えられている[13]。MutLαは弱いATPアーゼ活性を持つことが示されており、また非連続的なDNA鎖にさらにニックを導入するエンドヌクレアーゼ活性も持つ。これによって、EXO1によるミスマッチDNA鎖の5'→3'方向の分解が促進される[13]。MutLαの活性部位はPMS2サブユニットに位置する。PMS1とPMS2はMLH1との相互作用をめぐって競合する[13]。タンデムアフィニティ精製によって、PMS2と相互作用するタンパク質群が同定されている[13][14]。ヒトのPMS2は極めて低レベルで発現しており、細胞周期の強力な制御下には置かれていないと考えられている[15]。
p53やp73との相互作用
[編集]PMS2はp53やp73と相互作用することが示されている。p53が存在しない場合でも、PMS2が欠損した細胞と機能している細胞の双方でシスプラチン処理時の細胞周期のG2/M期チェックポイントでの停止は機能しているが、p53とPMS2の双方を欠損した細胞では抗がん剤に対する感受性が増大する[16]。PMS2はp53欠損細胞において細胞生存の媒介因子となっており、p53非依存的にDNA損傷応答を調節する[16]。PMS2とMLH1はミスマッチ修復に依存した形で、p73を介したアポトーシスに対抗することで細胞死からの保護を行う[16]。一方で、PMS2はp73と相互作用し、p73を安定化することでシスプラチン誘発性アポトーシスを促進する。シスプラチンはPMS2とp73の相互作用を促進し、この作用はc-Ablに依存している[12]。MutLα複合体はp73を損傷DNA部位へリクルートするアダプターとして機能している可能性があり、その中でPMS2はp73の活性化因子として作用している可能性がある。PMS2の過剰発現は、MLH1非存在下、p73とシスプラチンの存在下において、PMS2のp73に対する安定化作用によりアポトーシスを促進している可能性がある[12]。DNA損傷時には、p53はp21/WAF経路を介して細胞周期の停止を誘導し、MLH1とPMS2の発現によって損傷修復を開始する[11]。MutLα複合体はDNA損傷の程度のセンサーとして機能し、損傷が修復能力を超えた場合にはp73を安定化してアポトーシスを開始すると考えられている[11]。また、MLH1はMLH3やPMS1とも複合体を形成することができるため、PMS2の喪失は必ずしもMLH1の不安定性をもたらすわけではない[17]。
臨床的意義
[編集]変異
[編集]PMS2遺伝子はミスマッチ修復に関与するDNA修復タンパク質をコードする遺伝子である。PMS2遺伝子は7p22に位置し、15個のエクソンから構成される。7p22領域にはPMS2遺伝子ときわめて相同性が高い偽遺伝子が存在するが、エクソン11のコーディング領域の8つのアデノシンからなるリピート部分の配列が異なる。この偽遺伝子の配列が誤ってHNPCCやターコット症候群の原因変異とされている可能性がある[18]。
100,000種類のヒトがん試料の比較ゲノミクス解析からは、特にメラノーマにおいて、PMS2のプロモーター領域の変異が遺伝子変異量(TMB)と関係していることが示されている[19]。TMBは患者のがん免疫療法への応答性に関する信頼性の高い予測因子であり、TMBの高さはより良い治療成績と関係している[20]。
PMS2のようなDNAミスマッチ修復遺伝子のヘテロ接合性生殖細胞系列変異は、常染色体優性型遺伝疾患であるリンチ症候群(HNPCC)の原因となる。リンチ症候群の家系のうち、PMS2遺伝子に変異を有するのはわずか2%である[21]。PMS2と関係したリンチ症候群の症状が初めて出現する年齢は患者によって大きく異なり、23歳から77歳までの報告がある[22]。
稀な症例では、ホモ接合性の欠陥によってこの症候群が引きこされている。こうした症例では変異を両親から受け継いでおり、ターコット症候群もしくは体質性ミスマッチ修復欠損症候群(constitutional MMR deficiency、CMMRD)と呼ばれる[23]。2011年時点で、PMS2の両アレルの生殖細胞系列変異による脳腫瘍の患者が36例報告されている[23]。ターコット症候群の遺伝は優性と劣性の双方の場合があり、劣性遺伝はPMS2遺伝子の複合ヘテロ接合性変異を原因とする[24]。CMMRDの57家族のうち31家族はPMS2の生殖細胞系列変異を抱えている[25]。60人のホモ接合性もしくは複合ヘテロ接合性PMS2変異の保因者のうち19人では、消化器がんまたは腺腫がCMMRDの最初の症状であった[25]。偽遺伝子の存在によってPMS2遺伝子中の変異の同定に混乱が生じる場合があり、PMS2遺伝子変異の偽陽性判定がなされている場合がある[18]。
欠乏と過剰発現
[編集]PMS2の過剰発現は高変異性とDNA損傷抵抗性を引き起こす[26]。PMS2の欠乏もミスマッチ修復機能の低下を原因とする変異の増加によって遺伝的不安定性に寄与する[26]。Pms2-/-マウスではリンパ腫と肉腫が発生することが示されている。また、オスのPms2-/-マウスは不妊であることから、PMS2は精子形成に関与している可能性がある[7]。
正常な結腸における役割
[編集]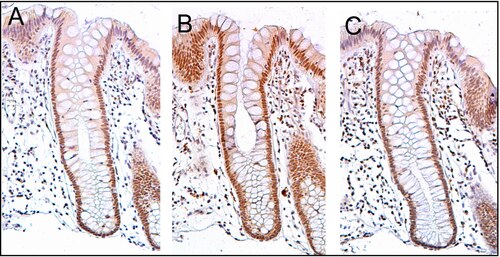
通常PMS2は、結腸の内側に並ぶ陰窩の腸細胞(吸収細胞)の核内で高レベルで発現している。新生物ではない正常な結腸上皮の陰窩では、PMS2、ERCC1、ERCC4(XPF)が関与するDNA修復が非常に活発に機能しているようである。PMS2の場合、正常な結腸上皮での高レベルの発現は陰窩の77%から100%で観察される[27]。
細胞は陰窩の底部で生み出され、陰窩の軸に沿って上方に移動し、数日後には結腸管腔へ脱落する[28]。陰窩の底部には5個から6個の幹細胞が存在する[28]。底部の幹細胞がPMS2を発現している場合、一般的にその陰窩の数千の細胞[29]全てでPMS2が発現している。このことはこの節の図のパネルAにおいて、陰窩内のほとんどの腸細胞がPMS2に対する免疫染色で褐色に染色されていることからも示される。正常な結腸上皮の各陰窩の数千の腸細胞では、ERCC1やERCC4でも同様の発現がみられる。この図ではヘマトキシリンによるDNAの対比染色も行われており、核が青灰色で示されている。粘膜固有層の細胞(陰窩の下部や周囲の細胞)の核の大部分はヘマトキシリンの青灰色を呈しており、PMS2、ERCC1、ERCC4をほとんど発現していない。
結腸がん
[編集]結腸がんの上皮由来の細胞の約88%、そしてがんから10 cm以内に近接して位置する(発がん可能性の高い「発がん素地」(field defect)内に位置する)陰窩の50%ではPMS2の発現が低下しているか欠損している[27]。
ミスマッチ修復欠乏もしくは欠損型として分類された腫瘍の大部分では、PMS2の発現の欠乏はその結合パートナーであるMLH1の欠損によるものである[30]。MLH1との結合によって、PMS2は安定化される[31]。MLH1の喪失がみられる散発性がん66症例のうち65症例では、プロモーターのメチル化によるエピジェネティックなサイレンシングが引き起こされていた。MLH1の発現がみられるもののPMS2が欠乏している16症例のうち、10症例は原因不明であったが、6症例ではPMS2にヘテロ接合型生殖細胞系列変異が発見されており、腫瘍でヘテロ接合性喪失が生じたものである可能性が高い。PMS2の変異によってPMS2の発現を欠いているものは119症例のうち6症例(5%)のみであった[27]。
ERCC1やERCC4との協調的機能
[編集]
発がん素地内の結腸陰窩でのPMS2の減少は、DNA修復遺伝子ERCC1やERCC4の発現低下と関係していることが多い。ERCC1やERCC4の欠乏はDNA損傷の蓄積を引き起こすと考えられており、こうした過剰なDNA損傷はアポトーシスを引き起こす[32]。しかしながら、これにPMS2の欠陥が加わることで、アポトーシスが阻害される[33][34]。そのため、ERCC1やERCC4が欠乏した際にはPMS2の欠乏が選択される可能性が高い。CHO細胞にDNA損傷刺激に繰り返し曝露した際の生存細胞に由来する5種類のクローンのうち、3種類ではPMS2に変異が生じていた[35]。
結腸がんへの進行
[編集]紫外光(DNA損傷を引き起こす)に曝露した際に、野生型CHO細胞と比較して、ERCC1、PMS2二重変異体細胞は7375倍、ERCC1単独変異細胞は967倍の変異頻度を示す[35]。このように、ERCC1とPMS2の双方が欠乏した結腸細胞ではゲノム不安定性が引き起こされる。PMS2とERCC4の双方に欠陥を有する細胞でも同様の状況が引き起こされると考えられる。こうした不安定性によってmutator phenotype(高変異表現型)が引き起こされることで結腸がんへの進行可能性が高まると考えられ[36]、結腸がんと関係した発がん素地にPMS2とERCC1(もしくはPMS2とERCC4)の双方が欠乏した細胞が存在することの説明となる。多くの種類のがんの根底には、DNA損傷に対して適切に応答して修復を行う能力の欠陥が存在している[37]。
出典
[編集]- ^ a b c GRCh38: Ensembl release 89: ENSG00000122512 - Ensembl, May 2017
- ^ Human PubMed Reference:
- ^ Mouse PubMed Reference:
- ^ “Mutations of two PMS homologues in hereditary nonpolyposis colon cancer”. Nature 371 (6492): 75–80. (Sep 1994). doi:10.1038/371075a0. PMID 8072530.
- ^ “Genomic organization of the human PMS2 gene family”. Genomics 30 (2): 195–206. (November 1995). doi:10.1006/geno.1995.9885. PMID 8586419.
- ^ “Entrez Gene: PMS2 PMS2 postmeiotic segregation increased 2 (S. cerevisiae)”. 2022年12月11日閲覧。
- ^ a b “PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance”. Proc. Natl. Acad. Sci. U.S.A. 107 (30): 13384–9. (12 July 2010). doi:10.1073/pnas.1008589107. PMC 2922181. PMID 20624957.
- ^ “Interactions of the DNA mismatch repair proteins MLH1 and MSH2 with c-MYC and MAX”. Oncogene 22 (6): 819–25. (February 2003). doi:10.1038/sj.onc.1206252. PMID 12584560.
- ^ a b “The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2”. Nucleic Acids Res. 29 (8): 1695–702. (April 2001). doi:10.1093/nar/29.8.1695. PMC 31313. PMID 11292842.
- ^ a b “The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer”. J. Biol. Chem. 274 (10): 6336–41. (March 1999). doi:10.1074/jbc.274.10.6336. PMID 10037723.
- ^ a b c “Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements”. Proc. Natl. Acad. Sci. U.S.A. 102 (13): 4813–8. (March 2005). doi:10.1073/pnas.0407069102. PMC 555698. PMID 15781865.
- ^ a b c “Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin”. Proc. Natl. Acad. Sci. U.S.A. 100 (5): 2420–5. (March 2003). doi:10.1073/pnas.0438031100. PMC 151356. PMID 12601175.
- ^ “PMS2 Gene”. The GeneCards Human Gene Database. Weizmann Institute of Science. 2022年12月11日閲覧。
- ^ “Cell cycle regulation of the human DNA mismatch repair genes hMSH2, hMLH1, and hPMS2”. Cancer Res. 57 (2): 206–8. (January 1997). PMID 9000555.
- ^ a b c “Increased sensitivity of p53-deficient cells to anticancer agents due to loss of Pms2”. Br. J. Cancer 87 (9): 1027–33. (October 2002). doi:10.1038/sj.bjc.6600599. PMC 2364320. PMID 12434296.
- ^ “Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation”. Cancer Res. 64 (14): 4721–7. (July 2004). doi:10.1158/0008-5472.CAN-03-2879. PMID 15256438.
- ^ a b “Polymorphisms in a pseudogene highly homologous to PMS2”. Hum. Mutat. 16 (6): 530. (December 2000). doi:10.1002/1098-1004(200012)16:6<530::AID-HUMU15>3.0.CO;2-6. PMID 11102987.
- ^ “Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden”. Genome Med. 9 (34): epub. (April 2017). doi:10.1186/s13073-017-0424-2. PMC 5395719. PMID 28420421.>
- ^ “Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers”. Mol. Cancer Ther. 16 (11): 2598–2608. (November 2017). doi:10.1158/1535-7163.MCT-17-0386. PMC 5670009. PMID 28835386.>
- ^ “PMS2 - PMS2 postmeiotic segregation increased 2 (S. cerevisiae)”. Genetics Home Reference. U.S. National Library of Medicine. 2022年12月20日閲覧。
- ^ “The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations”. Gastroenterology 135 (2): 419–28. (August 2008). doi:10.1053/j.gastro.2008.04.026. PMC 2759321. PMID 18602922.
- ^ a b “Childhood brain tumours due to germline bi-allelic mismatch repair gene mutations”. Clin. Genet. 80 (3): 243–55. (September 2011). doi:10.1111/j.1399-0004.2011.01635.x. PMID 21261604.
- ^ “Evidence for a recessive inheritance of Turcot's syndrome caused by compound heterozygous mutations within the PMS2 gene”. Oncogene 19 (13): 1719–1723. (March 2000). doi:10.1038/sj.onc.1203447. PMID 10763829.
- ^ a b “Paediatric intestinal cancer and polyposis due to bi-allelic PMS2 mutations: case series, review and follow-up guidelines”. Eur. J. Cancer 47 (7): 965–82. (May 2011). doi:10.1016/j.ejca.2011.01.013. PMID 21376568.
- ^ a b “Overexpression of the DNA mismatch repair factor, PMS2, confers hypermutability and DNA damage tolerance”. Cancer Lett. 244 (2): 195–202. (December 2006). doi:10.1016/j.canlet.2005.12.009. PMID 16426742.
- ^ a b c d e “Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer”. Genome Integr 3 (1): 3. (2012). doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ^ a b “Quantification of crypt and stem cell evolution in the normal and neoplastic human colon”. Cell Rep 8 (4): 940–7. (2014). doi:10.1016/j.celrep.2014.07.019. PMC 4471679. PMID 25127143.
- ^ “Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts”. Aging Cell 9 (1): 96–9. (2010). doi:10.1111/j.1474-9726.2009.00531.x. PMC 2816353. PMID 19878146.
- ^ “Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer”. Gastroenterology 128 (5): 1160–71. (2005). doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ^ “Steady-state regulation of the human DNA mismatch repair system”. J. Biol. Chem. 275 (24): 18424–31. (2000). doi:10.1074/jbc.M001140200. PMID 10747992.
- ^ “DNA damage-induced apoptosis”. Oncogene 23 (16): 2797–808. (2004). doi:10.1038/sj.onc.1207532. PMID 15077143.
- ^ “Functional role of DNA mismatch repair gene PMS2 in prostate cancer cells”. Oncotarget 6 (18): 16341–51. (2015). doi:10.18632/oncotarget.3854. PMC 4599273. PMID 26036629.
- ^ “Apoptotic function of human PMS2 compromised by the nonsynonymous single-nucleotide polymorphic variant R20Q”. Proc. Natl. Acad. Sci. U.S.A. 105 (37): 13993–8. (2008). doi:10.1073/pnas.0806435105. PMC 2528866. PMID 18768816.
- ^ a b “Highly elevated ultraviolet-induced mutation frequency in isolated Chinese hamster cell lines defective in nucleotide excision repair and mismatch repair proteins”. Cancer Res. 61 (1): 50–2. (2001). PMID 11196196.
- ^ “Human cancers express mutator phenotypes: origin, consequences and targeting”. Nat. Rev. Cancer 11 (6): 450–7. (2011). doi:10.1038/nrc3063. PMC 4007007. PMID 21593786.
- ^ “The DNA damage response: ten years after”. Mol. Cell 28 (5): 739–45. (2007). doi:10.1016/j.molcel.2007.11.015. PMID 18082599.
外部リンク
[編集]- FAQs on HNPCC Archived 2007-08-15 at the Wayback Machine. from the National Institute of Health
- GeneReviews/NCBI/NIH/UW entry on Lynch syndrome